







Research
Molecules in cells do not work in synchronization (work in essentially
heterogenous!), but do randomly.
The interaction periods of individual molecules are often less than a second.
In addition, the fraction of interacting molecules is frequently less than
10%. If we measure averaged behavior of many molecules in cells, we easily
miss such interaction, and it is very hard to unravel molecular mechanisms
in cells.
Therefore, we try to unravel the molecular mechanisms in cells by observing proteins and lipids at the level of single molecules (sometimes, at 10,000 frames/sec which is world’s fastest rate!), and by evaluating periods and frequencies of individual events.
Then, we perform statistical analysis by observing many events at the level of single molecules. (Of course, the results obtained by single-molecule imaging should be consistent with those by observation of multiple molecules.)
1. Unraveling membrane structures and their dynamics by high-speed super-resolution microscopy
In super-resolution single-molecule localization microscopy (SMLM), such as PALM and dSTORM, individual blinking molecules are detected, and their positions are determined over 500-10,000 frames to reconstruct a super-resolution image. Although the spatial resolution of SMLM is high (~20 nm), the acquisition of such images is time-consuming; SMLM is typically performed in fixed cells. By employing a high-speed single-molecule imaging technique, we succeeded obtained SMLM images within short acquisition times and generated super-resolution movies by linking sequential still images. This approach allows the visualization of membrane structural dynamics in living cells with a spatial resolution of 20 nm. For instance, Figure 1 shows images extracted every 5 seconds from a super-resolution movie of caveolae on the plasma membrane. The caveolae indicated by arrowheads are absent in the corresponding positions in adjacent frames, demonstrating that they form and disappear within this timescale.
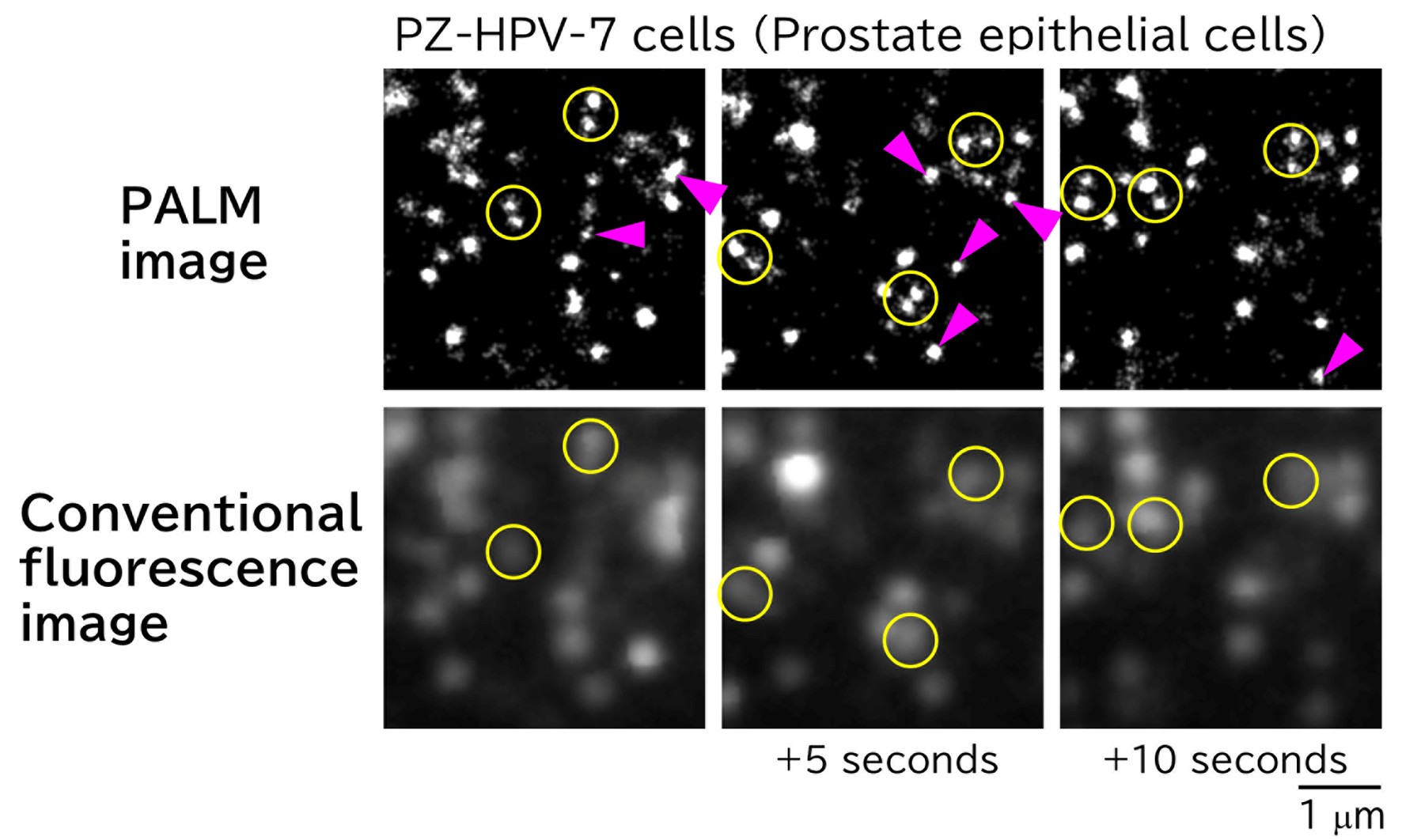
Figure 1. PALM images of caveolae in PZ-HPV-7 cells acquired every 5 seconds (top) and corresponding conventional fluorescence microscopy images (bottom). Each yellow circle in the PALM images contains two distinct caveolae, whereas only one is discernible by conventional fluorescence microscopy. A caveolae is visible at the magenta arrowhead; however, no caveolae are present at the same location 5 seconds earlier or later.
2. Mechanism of target cell modification by cancer cell-derived extracellular vesicles
In recent years, extracellular vesicles (EVs) have attracted considerable attention as pivotal mediators of intercellular communication. These vesicles carry molecular information, including nucleic acids and proteins, originating from the secreting cells. For instance, it has been proposed that when EVs derived from cancer cells are internalized by target cells in distinct organs, they create a microenvironment favorable for cancer metastasis. However, the detailed mechanism by which EVs bind to and are internalized by target cells remains largely unresolved.
We developed high-speed single-molecule localization microscopy techniques to address this issue. We discovered that EVs consist of distinct subtypes: for example, among EVs derived from PC3 cells, 60% contained only CD63, whereas 40% carried all three tetraspanin markers-CD63, CD9, and CD81. Moreover, as shown in Figure 2, we revealed that integrins α6β1 and α6β4 on cancer cell-derived EVs bind to laminin in the extracellular matrix on target cells, thereby facilitating EVs' adhesion. We further demonstrated that the laminin receptor integrin α6 in cancer cell-derived EVs enhances this binding through interaction with the tetraspanin protein CD151. In contrast, we found that talin and kindlin in EVs do not enhance the laminin-binding activity of integrins. Additionally, we identified that while integrin-laminin binding constitutes the primary adhesion mechanism, the ganglioside GM1 can also bind to laminin (Isogai et al., J. Cell Biol., 2025).
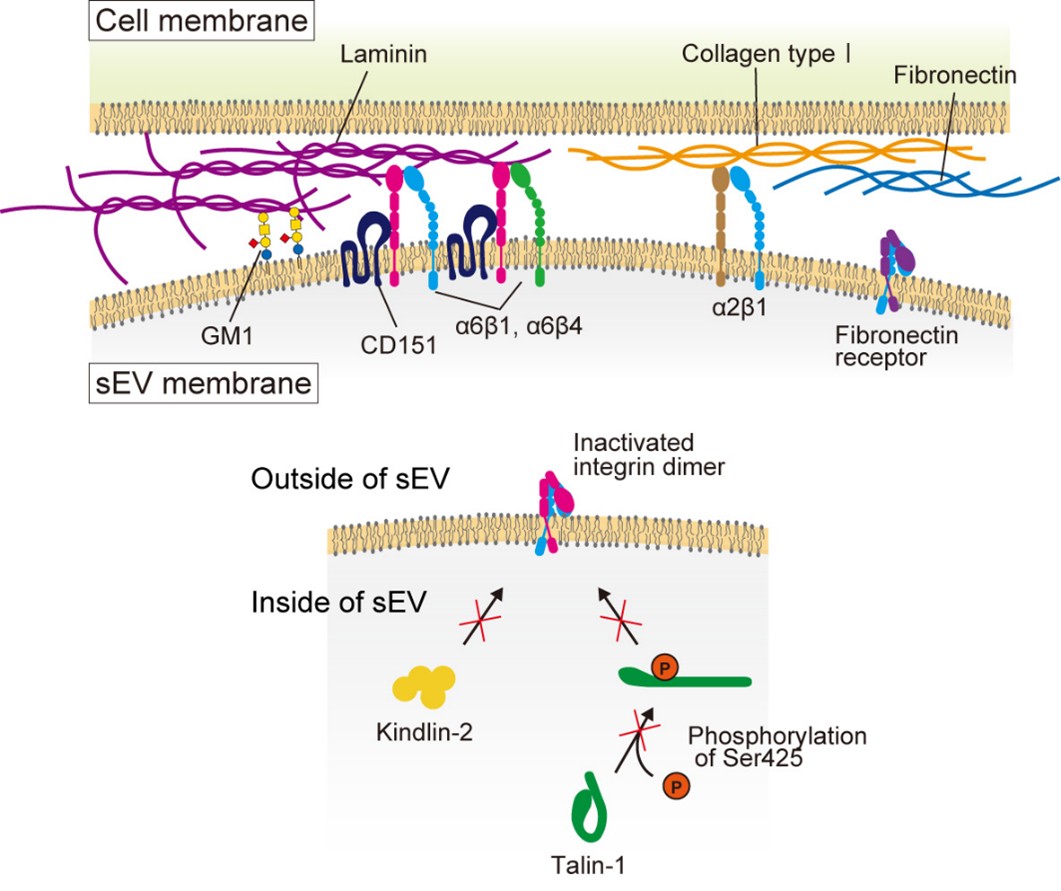
Figure 2. Integrin α6β1 and α6β4 on EV membranes bind to laminin on the target cell PM, enabling EV attachment to the target cell. In contrast, fibronectin receptors exhibit little binding activity. The tetraspanin CD151 associates with integrin α6β1, thereby enhancing its laminin-binding activity. Talin and kindlin within EVs do not contribute to integrin activation. Inhibitory molecules targeting laminin receptors associate through cholesterol. The ganglioside GM1 on EVs also exhibits laminin binding activity.
Next, to elucidate the mechanism underlying EV uptake by target cells, we simultaneously performed super-resolution movie observation of uptake membrane structures and single-particle tracking of sEVs containing the tetraspanin marker protein, CD63 (Figure 3). The results revealed that most sEVs are internalized via clathrin-independent endocytosis domains, visualized using LAMP-2C as a marker. Furthermore, we demonstrated that only CD63-positive EVs with low membrane fluidity as also internalized via caveolae (Hirosawa et al., Nat. Commun., 2025).
Surprisingly, the adhesion signaling of sEVs facilitates their uptake by target cells. Upon binding of sEVs to the target cell PM, integrin b1 becomes concentrated directly beneath the bound vesicles, inducing the formation of talin clusters. Subsequently, PLCg was activated via Src family kinases, leading to the hydrolysis of PI(4,5)P2 to IP3 within the inner leaflet of the target cell PM. The binding of IP3 to its receptor on the ER triggers an increase in intracellular calcium level. We further demonstrated that this calcium elevation enhances dynamin activity through calcineurin, thereby promoting EV endocytosis. Notably, this signaling cascade occurs extensively upon paracrine-type EV binding to recipient cells, but not during the autocrine-type interactions. (Hirosawa et al., Nat. Commun., 2025).
EVs hold great potential for applications in medicine, drug discovery, and diverse biotechnological fields, underscoring the critical importance of elucidating their underlying mechanisms. We will continue to advance our research through the use of cutting-edge imaging technologies.
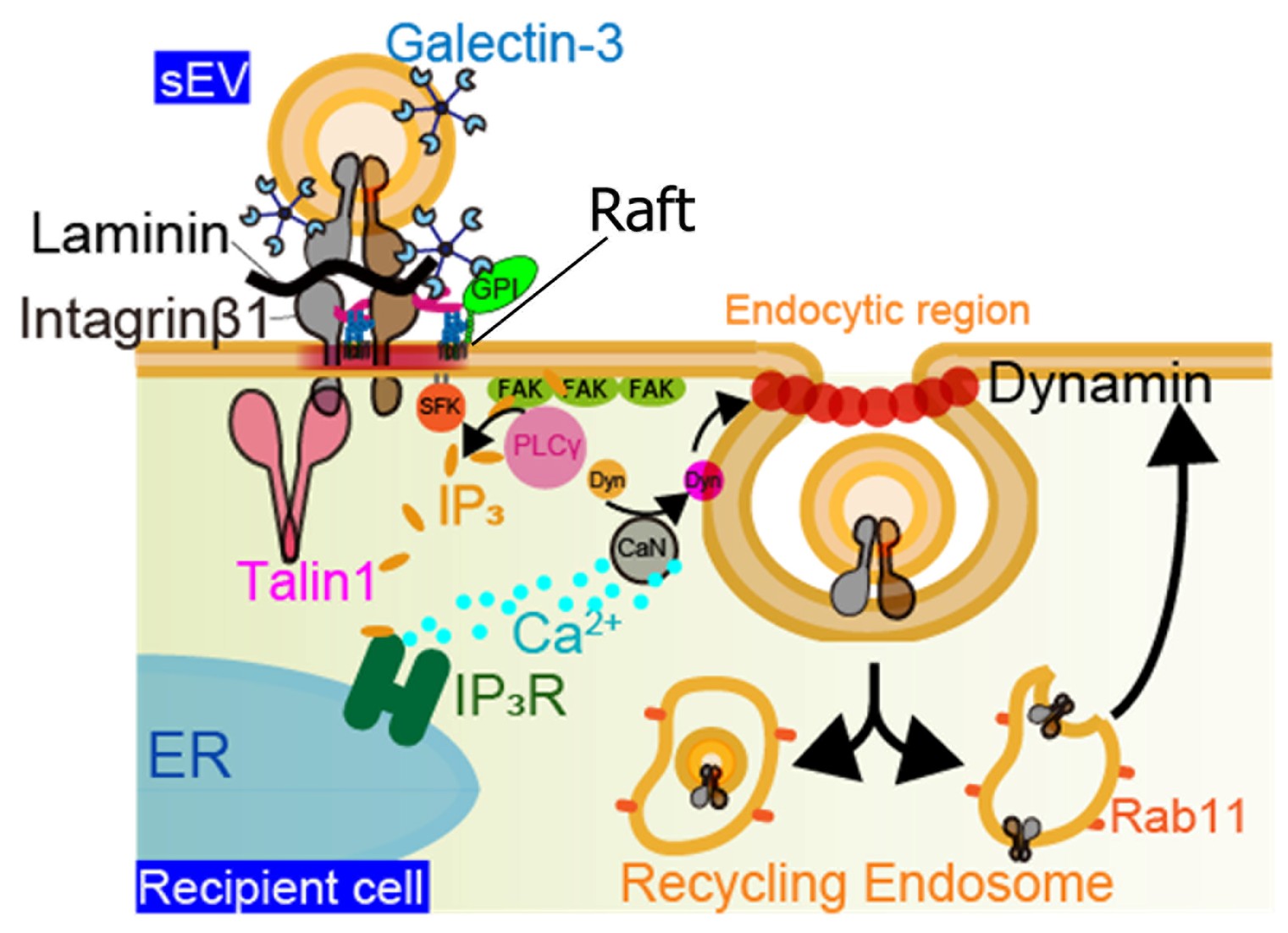
Figure 3. EVs bind to laminin on target cells, as shown in Figure 2. In addition, EVs are coated with galectin-3 (Gal-3), which bridges membrane proteins bearing LacNAc residues on both the cell PM and the EV membrane. LAMP-2C in the target cell PM interacts with Gal-3 on the EV surface,
mediating internalization through clathrin-independent endocytosis. In other words, this internalization is facilitated by Gal-3-LAMP-2C mediated binding of EVs to the target cell membrane. Integrin β1 accumulates directly beneath the bound EV on the target cell PM, followed by talin-1 clustering,
which triggers activation of Src family kinases (SFKs). Activated SFKs recruit PLCγ to FAK clusters, leading to the hydrolysis of PI(4,5)P2 into IP3 and eliciting an intracellular calcium response via IP3 receptors on the ER. The resulting elevation in intracellular Ca2+ enhances dynamin activity via calcineurin,
ultimately promoting clathrin-independent endocytosis and EV uptake via caveolae.
Press release 2025 [Isogai et al., 2025 J. Cell Biol.]
Press release 2025(1) [Hirosawa et al., 2025 Nat Commun.]
Press release 2025(2)
Press release 2025(3)
3. Unraveling the activation mechanism of the innate immune response molecule, STING
When cells are infected by DNA viruses, they elicit innate immune and inflammatory responses. The STING pathway serves as a critical defense mechanism against DNA virus infection; however, its abnormal activation is implicated in numerous pathological conditions, including innate immune disorders, neurodegenerative diseases, and cancer. Through collaborative research with the laboratory of Tomohiko Taguchi at Tohoku University, we elucidated the mechanism underlying STING activation using single-molecule super-resolution microscopy (Kemmoku et al., Nat. Commun., 2024).
We discovered that STING assembles into large clusters, each comprising more than 20 molecules on average, within cholesterol-rich lipid microdomains of the trans-Golgi network (TGN) through palmitoylation-dependent lipid modification. Moreover, the downstream kinase TBK1 is recruited to STING only after the formation of these large STING clusters (Figure 4).
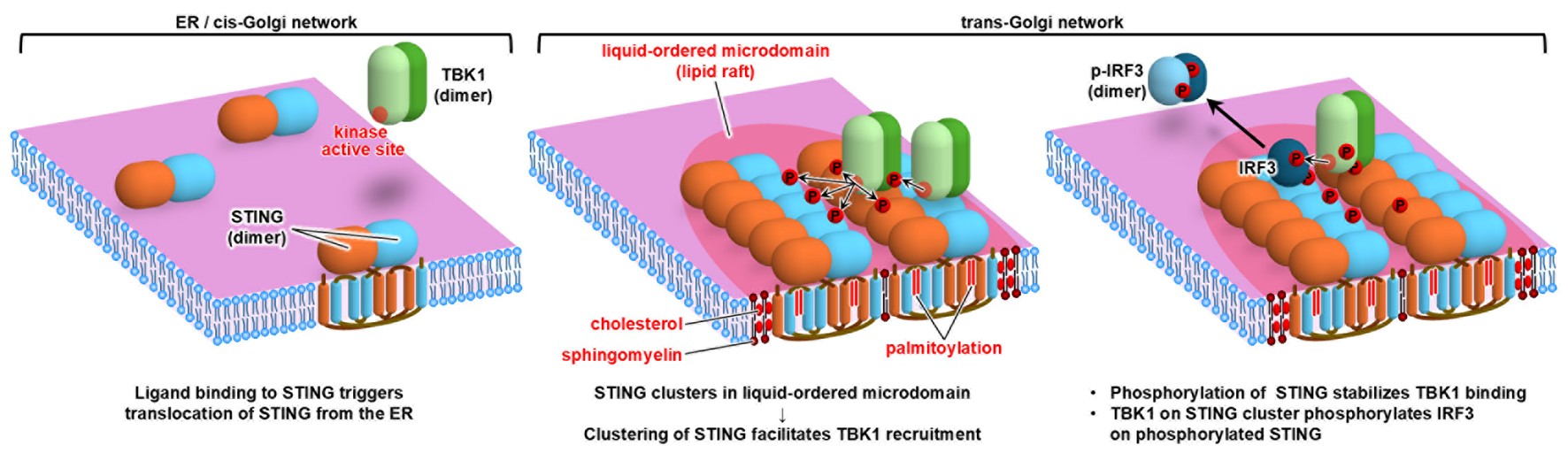
Figure 4. Before stimulation, STING exists as dimers on the ER (left). Upon ligand stimulation, STING translocates to the trans-Golgi network,
where it assembles into large clusters comprised of more than 20 molecules through lipid interactions between cholesterol and palmitoyl groups.
The downstream signaling kinase TBK1 is subsequently recruited to these high-density STING clusters.
Press release 2024(1) [Kemmoku et al. 2025 Nat. Commun.]
Press release 2024(2)
4. Unraveling of cell membrane structure, especially lipid raft structure and signal transduction
Lipid rafts have been drawing extensive attention as a signaling platform in cell membranes for about 30 years (Simons and Ikonen, Nature 1997). However, since rafts are too small to be observed by light microscopy, and molecules enter and go out of rafts very rapidly, rafts are still enigmatic. In early studies, nonionic mild detergents were used to extract “raft fraction”, or immunostaining methods were employed to detect rafts in cell membranes. However, it turned out that both methods tend to induce artifacts (Tanaka and Suzuki, Nature Methods, 2011). Therefore, to reduce the perturbation as small as possible, we observe the dynamic behavior of single molecules of representative raft molecules, GPI-anchored proteins (CD59, DAF, etc.) in living cell plasma membranes. We found that GPI-anchored proteins transiently form homodimers via protein-protein interactions, and them the homodimers are stabilized by cooperative raft-lipid interactions (Fig. 1, left). In other words, protein-protein interactions enhance raft-lipid interactions. We propose that the homodimers are one of the basic units to form greater rafts. At present, we are examining this model.
Furthermore, we are investigating how GPI-anchored proteins which lack transmembrane domain interact with signaling molecules in inner leaflet of cell membranes.
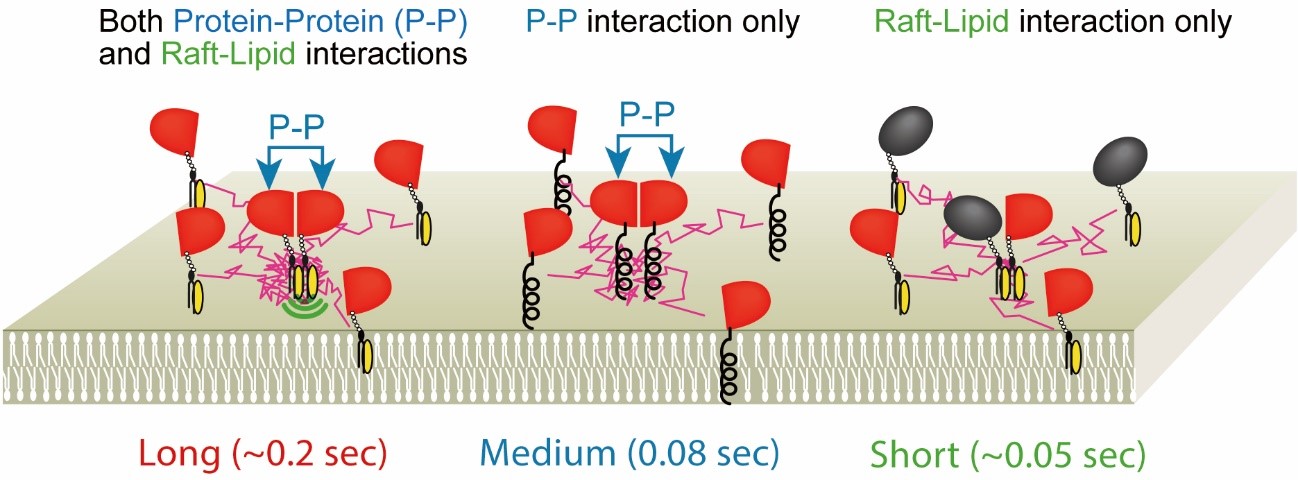
Fig.5. GPI-anchored protein, CD59, transiently (~200 ms) forms homodimers
via protein-protein interactions, which are stabilized by raft-lipid interactions.
The homodimer lifetimes were not elongated by raft-lipid interaction alone.
Press release 2010 [Tanaka and Suzuki et al., 2010 Nat. Methods]
Press release 2012 [Suzuki et al., 2012 Nat. Chem. Biol.]
5. Unraveling of dynamic behaviors of glycans in cell membranes
Glycans play very important roles in a variety of biological events. However, due to difficulty of synthesizing fluorescent probes, dynamic behaviors of glycans in living cell membranes have hardly been investigated. We tried to solve the issue and elucidate how glycans work in living cell membrane. Among glycans, we focus on “ganglioside” which is one of glycosphingolipid because it is a representative raft marker.
First, in collaboration with a group of Prof. Ando and Emeritus Prof. Kiso (G-CHAIN/Gifu University) we have recently developed four new fluorescent ganglioside probes (GM1, GM2, GM3, GD1b, Fig. 2) by using an entirely chemical method. These ganglioside probes act similarly to their parental molecules in terms of raft partitioning and binding affinity.
Since there have been no ganglioside fluorescent probes so far, dynamic behaviors of gangliosides have hardly been investigated in living cell plasma membranes. Using single fluorescent-molecule imaging, we have found that ganglioside probes dynamically enter and leave very tiny rafts at steady-state cells. Meanwhile, gangliosides also enter and leave rafts featuring CD59, a GPI-anchored protein. As the cluster size of CD59 increase (monomer→dimer→tetramer), the residency time of ganglioside probes in CD59 cluster was elongated (Fig. 3). In other words, we found that GPI-anchored proteins recruit other raftophilic lipids upon clustering, and stabilize the cluster rafts. This is also endorsed by another collaborative work with Prof. Murata (Osaka Univ.) and Prof Matsumori (Kyushu Univ.) in which new sphingomyelin probe we developed was used (Kinoshita and Suzuki et al., JCB, 2017).
Previous studies suggest that gangliosides regulate receptor activation in cell membranes, but the mechanisms in living cells have never known yet. At present, we are investigating how gangliosides regulate receptor clustering and activation, especially focusing on “glycan interactions”.
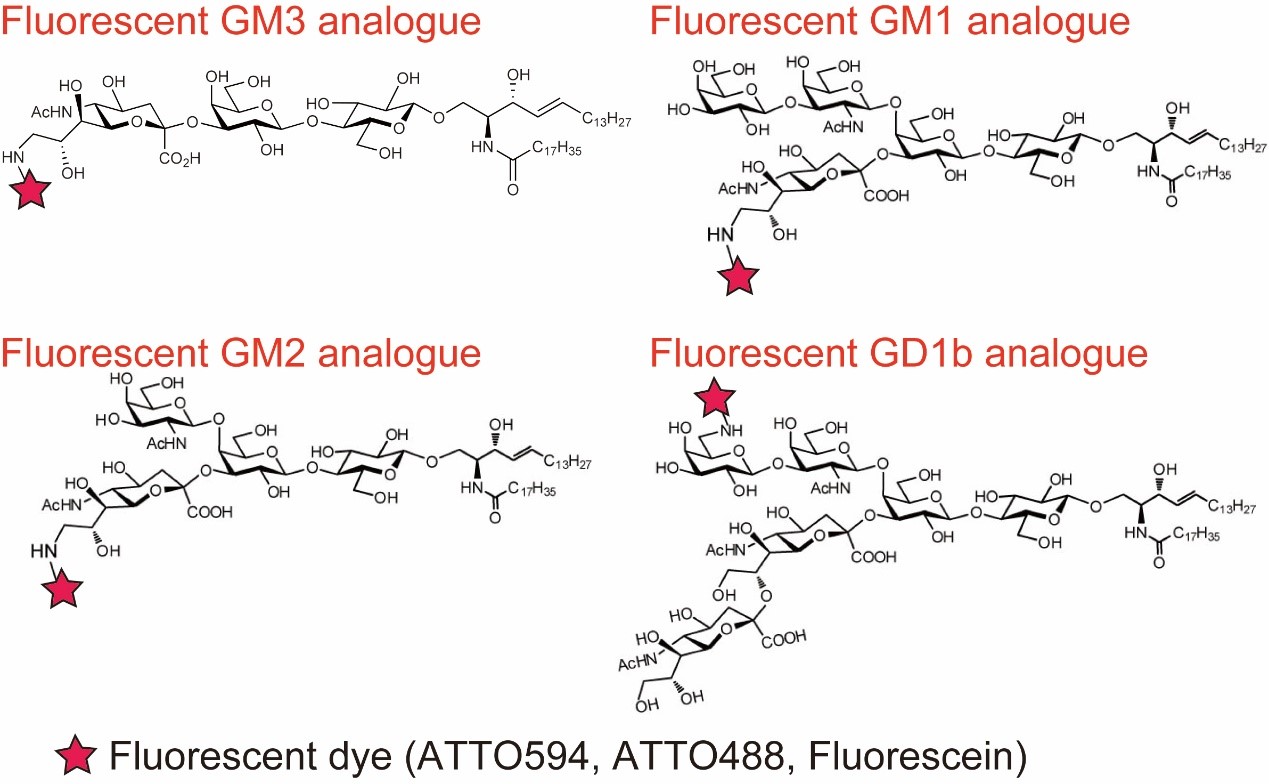
Fig. 6. Structure of fluorescent ganglioside probes which act similarly to their parental molecules in terms of raft partitioning and binding affinity
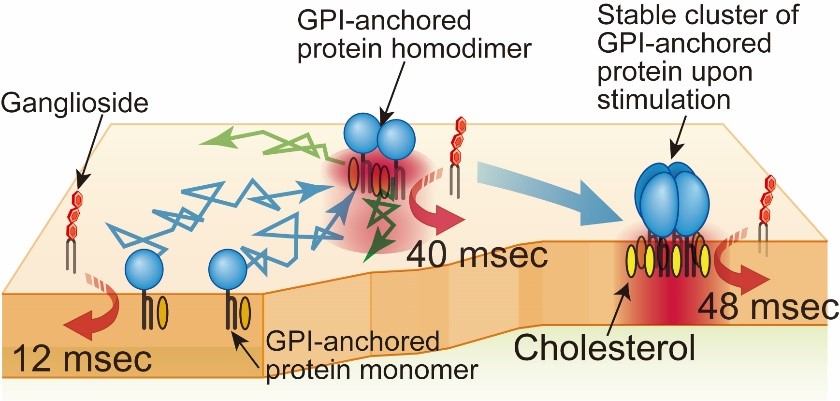
Fig. 7. Gangliosides were recruited to CD59 monomers, homodimers, and stabilized homotetramers for 12~48 ms, depending on cholesterol.
Press release 2016 [Komura, Suzuki and Ando et al., 2016 Nat. Chem. Biol.]
Press release 2017 [Kinoshita and Suzuki et al., 2017 J. Cell Biol.]
6.Unraveling of digital-like signaling system
Our results obtained by dual-color single-molecule imaging or single-molecule FRET of receptors and signaling molecules in living cell membranes indicate that signaling molecules are recruited to receptors for only less than a second, and suggest that signaling molecules are activated for very short terms (Fig. 4. Suzuki et al., 2007ab, J. Cell Biol.). On the other hand, cell bulk signals observed by western blotting of phosphorylation of signaling molecules continued for a few minutes to 20 minutes. Based on these results, we proposed a hypothesis of “digital-like signaling system” in which bulk signals continue over several minutes, but such bulk signals must be generated by the superposition of pulse-like individual events. In other words, the activation level of the bulk signal is determined by the superposition (integration) of the recruitment/activation of thousands of copies of the signaling molecule. The digital-like signaling system does not need complicated regulation and would be robust against noise. At present, we are testing this hypothesis.
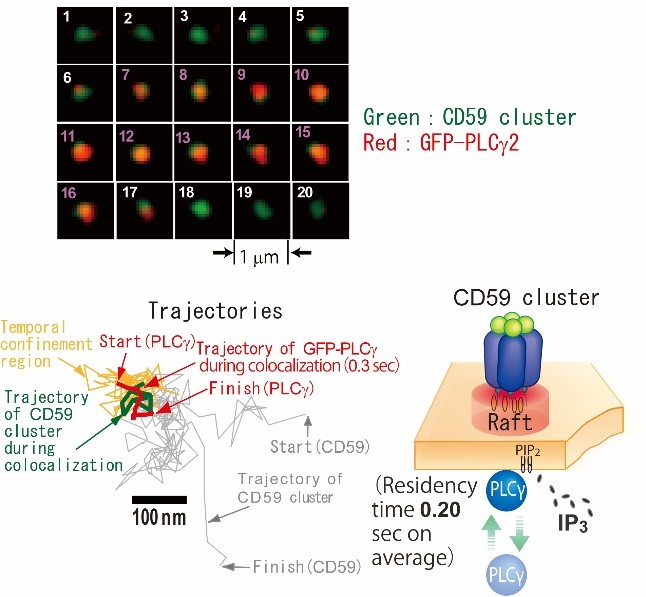
Fig. 8. Image sequence of dual-color single-molecule observation of PLCg and a CD59 cluster. PLCg (red) was recruited to a CD59 cluster (green) from frame 7 to 16 (Top). Trajectories of PLCg and CD59 cluster (Bottom left). Schematic representation of PLCg recruitment to CD59 cluster (Bottom right).
[Suzuki et al., 2007a, J. Cell Biol.] [Suzuki et al. 2007b, J. Cell Biol.]
Research
Research theme